 | |
МЕНЮ
- Главная
- Языкознание филология
- Финансовые науки
- Управленческие науки
- Товароведение
- Технология
- Теплотехника
- Теория организации
- Теория государства и права
- Таможенная система
- Схемотехника
- Строительство
- Страхование
- Статистика
- Религия и мифология
- Психология и педагогика
- Промышленность производство
- Медицинские науки
- Медицина
- Краеведение и этнография
- Компьютерные науки
- История
- Искусство и культура
- Информатика
- Инвестиции
- Издательское дело и полиграфия
- Зоология
- Журналистика
- Естествознание
- Деньги и кредит
- Делопроизводство
- Гражданское право и процесс
- Государство и право
- Геополитика
- Геология
- Геодезия
- География
- Военная кафедра
- Ветеринария
- Валютные отношения
- Бухгалтерский учет и аудит
- Ботаника и сельское хоз-во
- Биржевое дело
- Биология и химия
- Биология
- Безопасность жизнедеятельности
- Банковское дело
- Астрономия
- Астрология
- Архитектура
- Арбитражный процесс
- Административное право
- Авиация и космонавтика
- Карта сайта
Дипломная работа: Исследования свойств штамповой стали после термической обработки
2.2.1 Выявление микроструктуры
Для изучения микроструктуры образцов необходимо, чтобы их поверхность была специально приготовлена. Изготовление микрошлифа сводится к выполнению следующих операций: шлифование, полирование и травление.
Шлифование производилось на шлифовальной бумаге с постепенным переходом от бумаги марки № 12–3 с зернистостью от 125 до 20 мкм к бумаге марки М40–М5 с зернистостью от 28 до 3,5 мкм (ГОСТ 6456–75).
При переходе от одного номера зернистости к другому образец очищался от абразива и менялось направление шлифования на 90° для полного удаления всех рисок, образовавшихся во время предыдущей операции.
Полировка производилась на специальном полировальном станке, диск которого обтянут фетром, с помощью полировальной жидкости. После полировки образец был промыт водой и высушен фильтровальной бумагой.
Для выявления микроструктуры полированную поверхность микрошлифа подвергали травлению. Выбор состава травителя при этом зависел от конкретной поставленной задачи. В работе использовались следующие реактивы:
1) для выявления аустенитного зерна – пятипроцентный спиртовой раствор пикриновой кислоты, усиленный добавками 0,5–1% алкил-сульфата натрия;
2) для отделения реплик при электролитическом травлении применялся десятипроцентный спиртовой раствор азотной кислоты;
3) для выявления микроструктуры при электролитическом травлении – спиртовой раствор треххлористого железа и лимонной кислоты (0,5 г. FeCl3, 0,5 г. лимонной кислоты, 25мл. спирта).
2.2.2 Методика измерения твердости на приборе Роквелла
Измерение твердости производилось на приборе Роквелла с помощью алмазного конуса с углом при вершине 120° и радиусом закругления в вершине конуса 0,2 мм [12]. Суммарная нагрузка составила 1 500 Н (шкала С). Отсчет производился по черной шкале. Перед работой прибор проверялся с помощью эталона соответствующей твердости, после чего вносилась поправка в полученные значения твердости. Количество произведенных измерений не менее пяти для каждого образца.
2.2.3 Методика измерения микротвердости
Для определения микротвердости исследуемых материалов использовался микротвердомер ПМТ–3 с увеличением 480 крат, принцип работы которого заключается в том, что четырехгранная алмазная пирамида (с углом при вершине между противоположными гранями 136°) вдавливается в испытуемый металл под нагрузкой 2 Н.
Длина диагонали отпечатка определялась по формуле
Значения микротвердости определялись по формуле
![]() (1)
(1)
где P – нагрузка на пирамиду, г;
d – длина диагонали отпечатка, мкм.
При измерении необходимо учитывать неизбежный разброс полученных значений вследствие влияния соседних структурных составляющих с иной твердостью, различной толщины испытуемых элементов структуры, ошибки измерения и других причин. Для возможности статистической обработки результатов эксперимента на каждом образце проводили не менее шестидесяти замеров.
2.2.4 Методика определения глубины обезуглероженного слоя
Глубину обезуглероженного слоя определяют различными способами:
1) металлографическими методами, сущность которых заключается в определении глубины обезуглероженного слоя по структуре под микроскопом после соответствующей термообработки и травления;
2) методом замера термоэлектродвижущей силы на обезуглероженной и необезуглероженной поверхностях образца;
3) методом замера твердости;
4) химическим методом. [13]
В данной работе использовались методы замера твердости и микротвердости.
Метод замера твердости заключается в замере твердости образцов, подвергнутых термической обработке [13]. Замеры твердости производились на приборе Роквелла по ГОСТ 9013–59 непосредственно на поверхности образцов. Образец считали необезуглероженным, если его твердость соответствовала норме твердости, установленной по измерениям на необезуглероженной поверхности. В противном случае с поверхности снимался слой металла толщиной до 0,02 мм, и измерения повторялись. Количество проведенных измерений в каждом случае не менее пяти.
Метод замера микротвердости был реализован с помощью микротвердомера ПМТ–3. При измерениях учитывалось расстояние от обезуглероженной поверхности образца. Для возможности статистической обработки полученных данных было проведено по 10 замеров на каждом зафиксированном расстоянии от поверхности.
2.2.5 Выявление и определение величины аустенитного зерна
Выявление аустенитного зерна
Выявление зерна можно производить различными способами: методом окисления, методом цементации, методом нормализации, методом высокотемпературной металлографии [14].
В данной работе был использован метод окисления. Одна плоскость образцов заданной марки стали была последовательно отшлифована на грубой и тонкой наждачной бумаге. Приготовленные образцы помещены в печь обработанной стороной вверх. Заданный технологический режим находился под контролем. Известно, что кислород атмосферы печи, окисляя поверхность металла, наиболее интенсивно проникает по границам аустенитных зерен, и декорирует их. Образцы, охлажденные в воде и отшлифованные тем же номером наждачной бумаги с расчетом, чтобы на поверхности шлифа сохранилось 10–12% окалины (т.е. делался косой шлиф), были отполированы и потравлены [7].
В качестве травителя был применен пересыщенный водный раствор пикриновой кислоты, который сильнее воздействовал на участки, обогащенные кислородом. Избирательное действие пикриновой кислоты усиливалось добавками 0,5–1% алкил-сульфата натрия, а также травлением с той же добавкой в течение 40–50 минут при 80–85оС. По окончании травления шлиф был промыт холодной водой. Произведенное далее легкое полирование улучшило четкость выявленных границ, так как позволило удалить следы травления, окрашивающие поверхности зерен в разные цвета. [1]
Определение величины аустенитного зерна
Определение величины зерна может быть выполнено различными методами. В данной работе использованы следующие из них:
1) метод визуального сравнения видимых под микроскопом зерен с эталонной шкалой;
2) метод случайных секущих;
3) метод измерения длин хорд.
Метод определения величины зерна сравнением с эталонными шкалами. Величину зерна в работе определяли методом сравнения под микроскопом при увеличении 400 путем просмотра площади шлифа и сравнения видимых зерен с эталонной шкалой на увеличение 400 [14].
После просмотра десяти полей зрения шлифа, был установлен номер зерна, по которому можно дать количественные характеристики структуры, в частности, расчетный диаметр зерна.
Метод случайных секущих. Метод состоит в подсчете пересечении границ зерен случайной секущей. Такой секущей служит средняя линия окуляр – микрометра. Данным методом определяется средний условный диаметр – в случае равноосных зерен или количества зерен в 1 мм3 – в случае неравноосных зерен.
Для определения среднего размера зерен исследуемый образец был установлен на микроскоп и подсчитано количество зерен (число пересечений), укладывающихся на длине линейки окуляра. Увеличение микроскопа подобрано таким образом, чтобы на длине линейки окуляра укладывалось не менее 10 зерен.
Таких подсчеты были сделаны в пяти полях зрения для каждого образца.
Средний условный диаметр зерна (dср)
dср = (L/n) × Z, мм (2)
где L – суммарная длина всех отрезков в делениях окуляр-микрометра;
n – общее число зерен, пересеченных отрезками, длиной L;
Z – цена деления окуляр-микрометра для увеличения, при котором проводили подсчет пересечений зерен.
Для определения цены деления окуляр-микрометра вместо шлифа на столик микроскопа в работе устанавливали объект-микрометр, представляющий собой пластину, в центре которой имеется линейка с известной ценой деления (0,01 мм). После совмещения начальных делений обеих шкал объект-микрометра и окуляр-микрометра было подсчитано количество совпадающих делений. Цена деления окуляр-микрометра
Z = (с/а) × 0,01, мм (3)
где а – количество совпадающих делений окуляр-микрометра;
с – количество совпадающих делений объект-микрометра [8].
Метод измерения длин хорд. Метод основан на замере линейных размеров отрезков – хорд, отсекаемых в зернах прямыми линиями (линейкой окуляр-микрометра). Общее количество измерений зависит от однородности величины зерна, требуемой точности и достоверности результатов [14].
Замеры хорд были проведены по нескольким линиям в произвольном направлении на шлифе. Подсчитывалось количество длин хорд каждого размера по всем линиям.
Относительная длина зерен в процентах с определенной длиной хорды, соответствующей номеру группы
[li × ni/(li × ni)] × 100, % (4)
где li – длина хорды в i-ой группе;
ni – количество зерен с длиной хорды l;
(li × ni) – общая длина хорд всех групп.
По результатам измерений хорд построены гистограммы распределения хорд. Для того чтобы гистограмма правильно отражала закон распределения, весь массив хорд разбит на группы (интервалы), число которых не менее 8.[8]
Для статистической обработки экспериментальных данных и для нахождения точности фиксировалось общее количество зерен, подвергшихся измерениям. Количество произведенных замеров не менее 150 для каждого образца.
2.2.6 Методика нанесения покрытий
На образцы № 91, 30, 89 были нанесены покрытия из нитрида и оксинитрида титана. Покрытия наносились на установке для ионно-плазменной имплантации с титановым катодом (рис. 11). Перед нанесением покрытий образцы предварительно были отполированы и обезжирены в ультразвуковой ванне (Branson 3210).
Перед проведением работы в установке создавался вакуум 1×10-3 Па с помощью форвакуумного, а затем турбомолекулярного насоса.
Образец помещали в вакуумную камеру и обрабатывали в плазме аргона в течении десяти минут для очистки поверхности от загрязнений (давление аргона 0,5 Па).
Для нанесения покрытия в установку осуществлялся напуск азота и кислорода в различном соотношении и подавалось напряжение на титановый катод. Полученная в результате распыления катода плазма оседала на поверхности образца, создавая покрытие. Давление аргона и азота в камере составляло 6×10-2 Па; давление кислорода равнялось 6×10-2 Па и 2×10-2 Па для образцов 30 и 91 соответственно.
На образец также подавали отрицательное напряжение (2,5 В), необходимое для лучшего сцепления наносимого покрытия с поверхностью образца.
Напыление покрытия продолжалось в течение пяти минут. После завершения работы в установку напускали воздух из атмосферы и вынимали образец.
2.2.7 Определение износостойкости
Измерения износостойкости были выполнены с использованием трибометра МАТ GmbH. Применялся метод трения «шарика по диску». Шарик, диаметром 3 мм, был выполнен из твердого сплава ВК6. Перед испытаниями шарик и образец очищались в спиртовой ультразвуковой ванне (Branson 3210) в течении 5 минут для удаления поверхностных загрязнений. Далее образец наклеивался на подложку и помещался в трибометр, где его поверхность приходила в соприкосновение с нагруженным шариком (нагрузка на шарик 300 г.). Частота колебаний шарика составляла 3 Гц. Через заданное число циклов прибор автоматически отключался. В работе применялись два режима: 5 000 и 40 000 циклов.
Глубина и сечение следа трения измерялись на профилометре Dektak 8 000. Количество проведенных измерений сечения не менее пяти для каждого следа трения. По полученным данным рассчитывался вынесенный объем материала, по которому можно сравнительно оценить износостойкость. [15]
Также с помощью оптического микроскопа оценивался износ шарика, что позволило проверить достоверность полученных результатов. [15]
Для изучения карбидной фазы в данной работе применялась просвечивающая электронная микроскопия – метод анализа внутренней структуры и фазового состояния материалов. Анализ проводился с помощью электронного микроскопа ЭМ–200 при увеличениях 3 300 и 8 400.
Методика исследования заключается в получении тонких слепков (реплик), снятых с протравленной поверхности образца и отображающих его рельеф, и последующем просвечивании коротковолновыми электронными волнами для получения визуального изображения. [10]
Для возможности непосредственного наблюдения карбидов применялась углеродная реплика, которая была получена на вакуумной напылительной установке ВУП–4. Для отделения реплики использовалось электролитическое травление в десятипроцентном спиртовом растворе азотной кислоты при напряжении на катоде два вольта. Материал шлифа являлся анодом, катод был сделан из нержавеющей стали. Время отделения реплики порядка шести минут.
Определение размеров карбидов производилось непосредственно под электронным микроскопом при увеличении 8 400 путем сравнения с эталонной шкалой и последующим учетом увеличения микроскопа. Число измерений не менее 150. Весь размерный массив измерений был разбит на восемь групп, которые отражены в гистограммах распределения размеров карбидов.
2.2.9 Определение количественного соотношения структурных составляющих в сплаве
Объемные соотношения структурных составляющих определяют по их площади на плоскости шлифа. В основе метода лежит математический принцип Кавальери, согласно которому доля фазы (α) в объеме (V) сплава, на площади (F) шлифа и на секущей линии (L), равны друг другу
V = F = L = n/n. (5)
Широкое применение нашел линейный метод определения количественного соотношения структурных составляющих (метод Розиваля). По этому методу площади фаз вычисляют по длинам отрезков линейки, попавших на данную фазу [11].
В данной работе метод Розиваля был реализован на фотографиях, сделанных с электронного микроскопа.
Было определено число делений шкалы, попавших на карбидную фазу (а) в 10 полях зрения и вычислено объемное содержание карбидов по формуле
Vк = (aср/L) × 100%, (6)
где aср – средняя сумма отрезков шкалы в делениях линейки, попавших на карбиды;
L – длина всей шкалы (в делениях линейки), L = 100 мм.
2.2.10 Оценка ошибки измеряемых величин
Постановка исследования и способ отбора данных по выбранной методике должны обеспечивать надежность результатов и их точность, достаточную для решения конкретной задачи. Точность эксперимента определяется методами математической статистики с использованием нижеприведенных характеристик.
1. Математическое ожидание случайной величины Х (выборочное среднее)
![]() , (7)
, (7)
где Xi – i-ое значение измерения;
n – число измерений.
2. Среднее квадратичное отклонение Sх
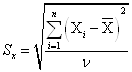 , (8)
, (8)
где ν – число степеней свободы, ν = n-1.
3. Дисперсия случайной величины Х
![]() . (9)
. (9)
4. Стандартное отклонение среднего результата
![]() . (10)
. (10)
5. Величина доверительного интервала ∆Х при заданной доверительной вероятности (Р = 0,95)
∆Х = t(ν, Р) × ![]() , (11)
, (11)
где t(ν, Р) – коэффициент Стьюдента, зависящий от числа произведенных измерений.
6. Относительная ошибка результата серии измерений
ε = ![]() . (12)
. (12)
Окончательный результат записывается в виде
Х = ![]() ± ∆Х. (13)
± ∆Х. (13)
Статистической обработке подвергали следующие измеренные величины:
1) значения твердости;
2) величины аустенитного зерна, измеренные методом хорд;
3) значения размеров карбидов, выпавших в реплику.
3.1 Влияние температуры закалки на структуру и твердость стали 4Х5МФ1С
Фотографии микроструктуры исследуемых образцов были получены с помощью микроскопа «EPIQUANT» после подготовки поверхности по вышеприведенной методике (см. п. 2.2.1). Травление осуществлялось электролитически при напряжении на катоде 2 В. В качестве травителя применялся спиртовой раствор треххлористого железа и лимонной кислоты.
Структура изучаемой стали представляет собой мартенсит, аустенит остаточный и карбиды (рис.12). Основная структура – мартенсит – плохо выявляется. Структуру необходимо просматривать при больших увеличениях, так как после закалки без перегрева зерна аустенита и кристаллы мартенсита очень малы.
С увеличением температуры нагрева под закалку, в конечном итоге, получаем более крупные мартенситные иглы, что можно наблюдать на фотографиях. Последнее обусловлено ростом действительного зерна аустенита при повышении температуры из-за стремления системы к минимуму свободной энергии. В свою очередь размер действительного зерна аустенита оказывает существенное влияние на дисперсность мартенсита и механические свойства стали в закаленном и отпущенном состоянии.
Общей для всех штамповых сталей тенденцией является большое содержание остаточного аустенита после закалки. Причем с повышением легированности твердого раствора понижаются температуры мартенситного превращения и возрастает количество аустенита. В комплекснолегированных сталях его количество может достигать 15–30% после обработки по оптимальным режимам и 60–80%после закалки с перегревом на 50–70оС. [4]
Концентрация углерода в мартенсите определяется химическим составом сталей и температурно-временными параметрами аустенизации. После обработки по принятым режимам (то есть на зерно не крупнее 8 балла) она составляет до 65–85% от общего содержания углерода в стали. При понижении температуры закалки фиксируемые в мартенсите содержания углерода уменьшаются, при этом его перераспределению в легированной стали сопутствует обеднение твердого раствора и легирующими элементами. Этот процесс наряду с выделением карбидов по границам зерен аустенита сопровождается также снижением пластичности, ударной вязкости и теплостойкости.
Твердость является важнейшим свойством инструментальных сталей. Она характеризует напряженное состояние, близкое к неравномерному всестороннему сжатию, и тем самым определяет сопротивление значительной пластической деформации и контактным напряжениям. С увеличением твердости в большинстве случаев возрастает износостойкость; увеличивается возможность получения более чистой и ровной поверхности как металла, обрабатываемого резанием или давлением, так и самого инструмента при его шлифовании или доводке; уменьшается налипание обрабатываемого металла на поверхность инструмента и т.д. В зависимости от состава стали и термической обработки твердость может изменяться в широких пределах.
В данной работе была измерена твердость закаленных образцов на приборе Роквелла. Количество измерений твердости составляет не менее пяти для каждого образца. Снятые экспериментальные данные были обработаны с использованием методов математической статистики. Экспериментальные данные представлены в таблице 6.
Таблица 6. Зависимость твердости стали 4Х5МФ1С от температуры закалки
| Маркировка образца |
Температура закалки, оС |
Твердость HRC |
Среднее квадратичное отклонение результата Sx |
Относительная ошибка ε, % | |||||
| № измерения | Среднее значение | ||||||||
| 1 | 2 | 3 | 4 | 5 | |||||
| 1 | 950 | 46 | 44 | 45 | 46 | 45 | 45 | 0,87 | 2,4 |
| 12 | 1 000 | 49 | 49 | 49 | 48 | 49 | 49 | 0,50 | 1,2 |
| 24 | 1 050 | 51 | 52 | 51 | 50 | 51 | 51 | 0,71 | 1,7 |
| 42 | 1 070 | 50 | 51 | 50 | 51 | 51 | 51 | 0,94 | 2,3 |
| 59 | 1 100 | 53 | 54 | 53 | 55 | 54 | 54 | 0,87 | 2,0 |
Исследования показали, что с увеличением температуры закалки увеличивается твердость (рис. 13), так как аустенит (мартенсит после охлаждения) становится более легированным за счет растворения карбидов при нагреве.
Страницы: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21
ИНТЕРЕСНОЕ
© 2009 Все права защищены. |