 | |
МЕНЮ
- Главная
- Языкознание филология
- Финансовые науки
- Управленческие науки
- Товароведение
- Технология
- Теплотехника
- Теория организации
- Теория государства и права
- Таможенная система
- Схемотехника
- Строительство
- Страхование
- Статистика
- Религия и мифология
- Психология и педагогика
- Промышленность производство
- Медицинские науки
- Медицина
- Краеведение и этнография
- Компьютерные науки
- История
- Искусство и культура
- Информатика
- Инвестиции
- Издательское дело и полиграфия
- Зоология
- Журналистика
- Естествознание
- Деньги и кредит
- Делопроизводство
- Гражданское право и процесс
- Государство и право
- Геополитика
- Геология
- Геодезия
- География
- Военная кафедра
- Ветеринария
- Валютные отношения
- Бухгалтерский учет и аудит
- Ботаника и сельское хоз-во
- Биржевое дело
- Биология и химия
- Биология
- Безопасность жизнедеятельности
- Банковское дело
- Астрономия
- Астрология
- Архитектура
- Арбитражный процесс
- Административное право
- Авиация и космонавтика
- Карта сайта
Учебное пособие: Аналитическая химия
Многие восстановители, такие как тиосульфаты, сульфиды, арсениты и др., титруют стандартным раствором йода. Часто прямое титрование невозможно по причине каталитического ускорения реакции окисления определяемого восстановителя кислородом воздуха. В этом случае прибегают к обратному титрованию: к раствору восстановителя добавляют избыток рабочего раствора йода, а его непрореагировавший остаток титруют вторым рабочим раствором тиосульфата натрия Nа2S2О3.
Сильные окислители определяют заместительным титрованием: к анализируемому раствору добавляют в избытке йодид калия и, если нужно, кислоту. При этом выделяется молекулярный йод в количестве, стехиометричном окислителю. Образовавшийся йод (заместитель) титруют рабочим раствором тиосульфата натрия. Прямое титрование сильных окислителей раствором Nа2S2О3 невозможно из-за образования разнообразных продуктов окисления.
Реакция йода с тиосульфатом имеет широкое применение в йодометрии:
I2 + Nа2S2О3 → 2NаI + Nа2S4О6
Она протекает быстро и стехиометрично при комнатной температуре в слабокислой среде или при рН < 9. При титровании сильнокислых растворов требуется интенсивное перемешивание, чтобы избежать разложения тиосульфата с образованием сернистой кислоты, которая реагирует с йодом, нарушая стехиометричность. В сильнощелочной среде протекают другие побочные реакции:
I2 + 2NаОН → NаIО + NаI + Н2О;
3NаIО → NаIО3 + 2NаI;
Nа2S2О3 + 4I2 + 10NаОН → 2Nа2SО4 + 8NаI + 5Н2О,
которые искажают результаты титрования.
Раствор тиосульфата натрия точно заданной концентрации нельзя приготовить путем растворения навески промышленного препарата Nа2S2О3 ∙ 5Н2О в мерной колбе. Состав препарата не отвечает точно указанной формуле, кроме того, свежеприготовленный раствор нестабилен. Основной причиной изменения его концентрации является деятельность тионовых бактерий, она усиливается при повышенной температуре и освещении. Наряду с этим тиосульфат натрия медленно реагирует с растворенной углекислотой и на свету − с кислородом воздуха:
Nа2S2О3 + Н2О + СО2 → NаНСО3 + NаНSО3 + S↓
2Nа2S2О3 + О2 → 2Nа2SО4 + S↓
Свежеприготовленный раствор тиосульфата натрия стерилизуют добавлением небольших количеств НgI2, хлороформа или амилового спирта, подщелачивают добавлением соды, защищают от света и выдерживают в течение 7-10 дней. Если раствор не стал мутным, то он устойчив и его стандартизуют.
Стандартизацию раствора тиосульфата натрия проводят с помощью стандартного вещества К2Сr2О7. При избытке КI и кислоты бихромат калия замещается стехиометричным количеством молекулярного йода q(К2Сr2О7) = q(I2) , который затем титруют тиосульфатом натрия q(I2) = q(Nа2S2О3) и, следовательно, q(Nа2S2О3) = q(К2Сr2О7).
Реакция замещения протекает медленно, для ее ускорения берут значительный избыток реагентов. Выделившийся йод мало растворим в воде, но при избытке йодид-ионов образуется более растворимое соединение трийдид-ион, понижающий упругость паров йода (его летучесть).
Окраска раствора йода при малой концентрации недостаточно интенсивна для точного определения конца титрования. В качестве чувствительного индикатора применяют свежеприготовленный коллоидный раствор крахмала, который образует с молекулярным йодом адсорбционное соединение интенсивно-синего цвета. При титровании йода тиосульфатом индикатор вводят в конце титрования, когда концентрация йода мала. Адсорбционное соединение, образующееся при большой концентрации йода, медленно реагирует с тиосульфат-ионом и это приводит к повышенному расходу тиосульфата натрия. Стандартный раствор тиосульфата натрия проверяют каждые 1-2 месяца.
3.4. Стандартизация раствора тиосульфата натрия
В основе определения лежат следующие реакции:
K2Cr2O7 + 6KI + 7H2SO4 = Cr2(SO4)3 + 4K2SO4 + 7H2O + 3I2
станд. в-во
I2 + 2Na2S2O3 = Na2S4O6 + 2NaI
раб.р-р
По уравнению реакции: n(Na2S2O3) = 6n(K2Cr2O7) = n(1/6 K2Cr2O7)
Раствор бихромата калия Eº(Cr2O72−/2Cr3+) = 1.33 В готовят по точной навеске в мерной колбе. Расчет интервала навески проводят согласно методике (см.раздел 2.2.1) исходя из приблизительной концентрации раствора Na2S2O3 = 0,05 мг-экв/мл.
Методика анализа
В колбу для титрования помещают 1 г йодистого калия, цилиндром отмеряют 5 мл 4 н серной кислоты и отмеряют пипеткой аликвотную часть приготовленного раствора бихромата калия.
Смесь в колбе для титрования перемешивают круговыми движениями до растворения йодида калия и для завершения реакции помещают на 5 мин в темное место.
Затем выделившийся йод титруют раствором тиосульфата натрия до получения светло-желтой (соломенной) окраски, к раствору добавляют пипетку раствора крахмала и продолжают титрование до исчезновения синей окраски.
По результатам трех титрований вычисляют средний объем Vср(Nа2S2О3), мл и вычисляют концентрацию раствора. Стандартизованный раствор тиосульфата натрия используют для определения точной концентрации рабочего раствора йода.
3.5. Стандартизация раствора йода
В основе определения лежит реакция:
I2 + 2Na2S2O3 = Na2S4O6 + 2NaI
раб.р-р 2-ой раб.р-р
По закону эквивалентов: q(I2) = q(Nа2S2О3)
Методика анализа
В колбу для титрования из бюретки последовательно отмеряют 4,0; 5,0 и 6,0 мл раствора йода и титруют стандартизованным раствором тиосульфата натрия до появления светло-желтого окрашивания раствора, затем добавляют пипетку раствора крахмала и продолжают титрование до исчезновения синей окраски.
На основании полученных результатов по закону эквивалентов рассчитывают концентрацию раствора йода:
N(Nа2S2О3) V(Nа2S2О3) = N(I2) V(I2)
Лабораторная работа №7
"Определение гексацианоферрат(II) калия"
Сущность определения заключается в способе обратного титрования: к аликвотной части задачи добавляют отмеренный избыток раствора I2. Его эквивалентное количество взаимодействует с определяемым веществом К4[Fе(СN)6], а остаток титруют вторым рабочим раствором
Nа2S2О3, т.е. q(К4[Fе(СN)6]) = q(I2) − q(Nа2S2О3).
Таким образом, данное определение основывается на следующих реакциях:
К4[Fе(СN)6] + I2 = К3[Fе(СN)6] + 2КI + I2
отмерен. остаток
избыток
I2 + 2Na2S2O3 = Na2S4O6 + 2NaI
остаток 2-ой раб.р-р
Методика анализа
Полученную задачу разбавляют в мерной колбе до метки дистиллированной водой и тщательно перемешивают. Аликвотную часть задачи пипеткой переносят в колбу для титрования и добавляют из бюретки 10 мл рабочего раствора йода. Так как отмеряют большой объем раствора йода, то необходимо отбирать пробу не спеша, чтобы исключить возможную ошибку натекания. Содержимое колбы перемешивают и помещают в темное место на 5 мин для завершения реакции. Избыток йода титруют рабочим раствором тиосульфата натрия, добавляя крахмал в конце титрования. Титрование заканчивают в момент исчезновения синей окраски. Объем тиосульфата натрия, ушедший на титрование пробы, записывают в рабочую тетрадь. Определение повторяют трижды и рассчитывают среднее значение объема Vср(Na2S2O3), мл. Результаты анализа выражают в граммах содержания К4[Fе(СN)6] в задаче:
V(I2) ∙ N(I2) Vср(Na2S2O3)∙ N(Na2S2O3)
m(К4[Fе(СN)6]) =10 ∙│ − -│∙Мэ(К4[Fе(СN)6])
1000 1000
4. МЕТОДЫ ОСАДИТЕЛЬНОГО ТИТРОВАНИЯ
Методы осадительного титрования основаны на реакциях, сопровождающихся образованием малорастворимых соединений. Они классифицируются в зависимости от применяемого рабочего раствора:
нитрата серебра (АgNО3) − аргентометрия;
нитрат ртути (I) (Нg2(NО3)2) − меркурометрия;
нитрат ртути (II) (Нg(NО3)2) − меркуриметрия;
роданид калия (аммония) КSСN (NН4SСN) − роданометрия.
4.1. АРГЕНТОМЕТРИЯ
Раствор нитрата серебра готовят из точной навески перекристаллизованной соли АgNО3 (х.ч.). Но так как титр раствора АgNО3 изменяется при хранении, то его необходимо периодически проверять. Точную концентрацию раствора АgNО3 устанавливают по стандартному веществу NаСl (х.ч.).
4.2. Стандартизация раствора нитрата серебра методом Мора
Метод Мора − это способ фиксирования точки эквивалентности с применением индикатора хромат калия (К2СrО4). Этот метод применим только для титрования в нейтральной или слабощелочной среде (рН = 6,5 ÷ 10,3), так как образующийся в результате реакции осадок Аg2СrО4 растворяется в кислой среде из-за ассоциации хромат-анионов, а в щелочной среде образуется осадок Аg2О. Ограничение метода связано с возможным присутствием в растворе мешающих ионов, образующих с хромат-анионами или с катионами серебра осадки (Ва2+, Рb2+, Вi3+, РО43−, АsО43−, С2О42− и др.), восстановителей, способных восстановить СrО42− до Сr3+ комплексообразователей ионов серебра.
Метод Мора применим для определения серебра, хлоридов и бромидов. Определение йодидов и роданидов этим методом не производится, так как образующиеся осадки АgI и АgSСN адсорбируют ионы СrО42−, что мешает фиксированию точки эквивалентности.
В основе метода лежат реакции:
NаСl + АgNО3 → АgСl↓ + NаNО3
раб.р-р белый
АgNО3 + К2СrО4 → Аg2СrО4↓ + КNО3
изб.капля индикатор кирп.-кр.
По уравнению реакции и с учетом закона эквивалентов: q(NаСl) = q(АgNО3).
Для проведения анализа необходимо рассчитать интервал навески NаСl, предполагая, что на титрование аликвотной части этой навески расходуется от 5 до 7 мл рабочего раствора нитрата серебра. Концентрация раствора АgNО3 ~ 0,05 г-экв/л.
Рассчитанную навеску NаСl взвешивают на аналитических весах, переносят в мерную колбу, растворяют, доводят до метки и тщательно перемешивают.
Методика анализа
В колбу для титрования переносят аликвотную часть стандартного раствора NаСl, добавляют 1 капля 5 % раствора К2СrО4 и титруют при энергичном перемешивании раствором АgNО3 до перехода желтой окраски суспензии в розовую (желтую окраску раствору придают хромат-ионы). Титрование повторяют трижды и рассчитывают значение Vср(АgNО3), мл. В процессе работы все отходы раствора АgNО3 сливаются в специальную емкость для отходов солей серебра. Растворы солей серебра вызывают химические ожоги, поэтому необходимо соблюдать осторожность при работе с ними. По данным титрования рассчитывают точную концентрацию раствора АgNО3 с учетом закона эквивалентов:
1000 mн(NаСl)
N(АgNО3) = ----------------------
10 Мэ(NаСl) Vср(АgNО3)
4.3. Стандартизация раствора роданида аммония
Концентрацию раствора роданида аммония устанавливают по стандартизованному раствору АgNО3 методом Фольгарда. В этом методе точка эквивалентности фиксируется с помощью индикатора − раствора железоаммонийных квасцов (Fе(NН4)(SО4)2 12Н2О), содержащим небольшое количество концентрированной азотной кислоты для подавления гидролиза. При избытке Fе3+ и малой концентрации SСN− наиболее устойчив комплексный анион [Fе(SСN)6]3−, окрашивающий раствор в интенсивно красный цвет.
Метод Фольгарда основан на реакции осаждения ионов серебра роданид-ионами. Растворы солей серебра титруют раствором NН4SСN в присутствии индикатора. Если точка эквивалентности не достигнута, то концентрации ионов SСN− мала и образование комплексов [Fе(SСN)6]3− не происходит. При небольшом избытке ионов SСN− комплекс образуется и раствор над осадком приобретает оранжево-красную окраску:
АgNО3 + NН4SСN → АgSСN↓ + NН4NО3
раб.р-р 2-ой раб.р-р белый
NН4SСN + Fе3+ = (NН4)3[Fе(SСN)6]
изб.капля индик. р-р оранжево-кр.
По уравнению реакции с учетом закона эквивалентов: q(АgNО3) = q(NН4SСN).
Метод Фольгарда (прямое титрование) применяется для определения серебра. Для определения бромидов и хлоридов применяют способ обратного титрования с использованием двух рабочих растворов: NН4SСN и АgNО3.
Методика анализа
В коническую колбу последовательно из бюретки отмеряют 4,0; 5,0 и 6,0 мл стандартизованного раствора АgNО3, цилиндром вносят 2 мл 6 н НNО3, разбавляют раствор, смывая все капли растворов внутрь колбы дистиллированной водой и добавляют 1 каплю раствора железо-аммонийных квасцов.
Титрование проводят раствором роданида аммония, постепенно приливая и резко встряхивая содержимое колбы, отделяя раствор от осадка. Вблизи точки эквивалентности роданид аммония добавляют по каплям, каждую каплю тщательно перемешивают и фиксируют изменение цвета раствора над осадком.
Титрование проводят трижды и рассчитывают три значения концентрации раствора NН4SСN по которым вычисляют точную концентрацию раствора роданида аммония с учетом закона эквивалентов:
N(NН4SСN) V(NН4SСN) = N(АgNО3) V(АgNО3)
Лабораторная работа №9
"Определение бромида калия методом Фольгарда"
В основе определения лежат реакции:
КВr + АgNО3 → АgВr↓ + КNО3 + АgNО3
отмер. изб. желт. остаток
АgNО3 + NН4SСN → АgSСN↓ + NН4NО3
остаток 2-ой раб.р-р белый
NН4SСN + Fе3+ = (NН4)3[Fе(SСN)6]
изб.капля индик. р-р оранжево-кр.
Расчет содержание бромида калия осуществляется по уравнениям реакций с учетом закона эквивалентов:
q(КВr) = q(АgNО3) − q(NН4SСN)
Методика анализа
В мерную колбу получают задачу, доводят до метки дистиллированной водой и тщательно перемешивают.
В коническую колбу для титрования пипеткой переносят аликвотную часть исследуемого раствора, с помощью бюретки отмеряют 10 мл раствора АgNО3, исключая возможную ошибку натекания, цилиндром отмеряют 5 мл 6 н раствора НNО3 и разбавляют раствор, смывая дистиллированной водой со стенок все капли раствора внутрь колбы для титрования.
Полученную смесь перемешивают и ждут, пока бромид серебра осядет на дно. Добавляют 1 каплю насыщенного раствора железо-аммонийных квасцов. Избыток АgNО3 титруют раствором роданида аммония до перехода окраски раствора над осадком в красно-розовый.
В рабочую тетрадь записывают объем NН4SСN, ушедший на титрование.
Определение повторяют трижды и вычисляют значение среднего объема роданида аммония Vср(NН4SСN), мл.
Результаты анализа выражают в граммах содержания КВr в задаче:
V(АgNО3) N(АgNО3) Vср(NН4SСN) · N(NН4SСN)
m(КВr) = 10 · │---------------------------- − ---------------------│·Мэ(КВr)
1000 1000
Лабораторная работа №10
"Потенциометрическое определение хлорид-, бромид- и йодид-ионов при совместном присутствии"
Потенциометрическое титрование, основанное на реакциях осаждения, применяется для определения серебра, а так же хлоридов, бромидов, йодидов, роданидов и сульфидов. В качестве титрующего раствора применяется рабочий раствор АgNО3. Электродом определения служит серебряный электрод − металлический индикаторный электрод I-го рода. Электродом сравнения служит хлорсеребряный электрод − металлический индикаторный электрод II-го рода.
Потенциометрический метод позволяет определить концентрации нескольких ионов при их совместном присутствии при условии, что отношение растворимостей образующихся при титровании серебряных солей должно быть не менее 40.
При добавлении раствора АgNО3 к исследуемому раствору, содержащему смесь хлорид-, бромид- и йодид-ионов, образуются осадки малорастворимых солей серебра. Значения произведения растворимости галогенидов серебра различно. ПРАgI = 10−16; ПРАgВr = 10−13; ПРАgСl = 10−10. При титровании вначале образуется осадок, у которого ПР имеет наименьшее значение (АgI), затем выпадает осадок АgВr, и последним осаждается АgСl. Осаждение АgI будет происходить до тех пор, пока произведение концентраций Аg+ и Вr− будет меньше значения их произведения растворимости: С(Аg+) · С(Вr−) < ПРАgВr, где С(Вr−) − концентрация ионов брома в исследуемом растворе. Осадок бромида серебра начнет образовываться, когда концентрация ионов серебра в растворе настолько возрастет, что С(Аg+) С(Вr−) = ПРАgВr.
Концентрация ионов йода в точке эквивалентности зависит от концентрации ионов брома в исходном растворе. Чем выше концентрация ионов, тем больше будет ошибка при титровании. Если концентрации ионов брома и йода одинаковы, то ошибка титрования минимальна.
Аналогично в точке эквивалентности при осаждении бромида серебра ошибка титрования будет зависеть от концентрации ионов хлора в исходном растворе. Ошибка при титровании в точке эквивалентности будет мала, если концентрация ионов хлора в исследуемом растворе будет в 5 раз меньше концентрации ионов брома.
Методика анализа
В мерную колбу получают раствор задачи, в которую предварительно для уменьшения адсорбции ионов галогенидов на поверхности осадка было введено 25 мл 7 % раствора нитрата бария. Объем в колбе дистиллированной водой доводят до метки и тщательно перемешивают.
В стаканчик для титрования пипеткой вносят две аликвотные части раствора задачи, объем в стаканчике доводят до 50 мл дистиллированной водой и опускают туда серебряный электрод. При помощи солевых мостиков, содержащих насыщенные растворы КСl и КNО3 соединяют реакционный сосуд с хлорсеребряным электродом.
Схема гальванического элемента:
(−) Аg │АgI, I−, Вr−, Сl− ║ КNО3 ║ КСl ║ КСl, АgСl│ Аg (+)
серебр.эл-д исслед.р-р хлорсеребр.эл-д
Включают магнитную мешалку, отрегулировав скорость вращения, и измеряют ЭДС гальванического элемента с помощью иономера. ЭДС цепи определяется: Е = φ°АgНаl − φ°хлорсер. − 0,058 lg СНаl-
Теоретические основы потенциометрического титрования рас-сматриваются в разделе 6. Титрование осуществляют рабочим раствором АgNО3 с установленным титром, прибавляя его по 0,2 мл. Результаты измерения вносят в таблицу (см. раздел 6) и по полученным данным на миллиметровой бумаге строят интегральную зависимость Е = f(Vраб.р-ра). По кривой определяют примерное положение точек эквивалентности. Для нахождения эквивалентных объемов строят дифференциальную кривую в координатах ∆Е/∆V от V'. Интегральная и дифференциальная кривые должны быть построены в одном масштабе. Дифференциальная кривая будет иметь три максимума. Проведя перпендикуляр от этих максимумов на ось абсцисс, находят значения трех эквивалентных объемов V1', V2' и V3' с точностью до 0,02 мл. Результаты анализа выражают в граммах галогенид-ионов в задаче:
V1’ N(АgNО3) · Мэ(I) · Vк
m(I−) = ---------------------------------
1000 2Vп
( V2' − V1') N(АgNО3) · Мэ(Вr) · Vк
m(Вr−) = --------------------------------------------
1000 2Vп
( V3' − V2') · N(АgNО3) · Мэ(Сl) · Vк
m(Сl−) = -------------------------------------------- , где
1000 2Vп
V1', V2', V3' − эквивалентные объемы, мл;
Vк − объем колбы, мл;
Vп − объем пипетки, мл.
5. МЕТОД КОМПЛЕКСОНОМЕТРИЧЕСКОГО ТИТРОВАНИЯ
Комплексонометрия − раздел титриметрического анализа, основанный на реакциях комплексообразования между определяемым ионом металла (М) и титрантом (комплексоном).
Катионы металла (М), содержащие вакантные d-орбитали, реагируют с донорами электронной пары − лигандами (L), образуя координационные соединения − хелаты. Хелатообразующий лиганд с двумя донорными группами, участвующими в образовании координационной связи, называется бидентатным; лиганд с тремя донорными группами − тридентатным и т.д.
Хелатообразование всегда протекает в одну стадию, тогда как при образовании комплекса образуется несколько промежуточных соединений.
В аналитических целях реакцию комплексообразования можно использовать, только если образуется комплекс типа МL состава 1:1, в котором к каждому металла присоединен один лиганд, поскольку для таких систем характерно наибольшее изменение значения рМ вблизи точки эквивалентности.
Наиболее широкое применение в комлексонометрии нашли комплексоны. Это полиаминокарбоновые кислоты, образующие с ионами металлов комплексные соединения: этилендиаминтетрауксусная кислота (ЭДТУ) − комплексон II и ее двунатриевая соль (ЭДТА), которую называют комплексон III или трилон Б:
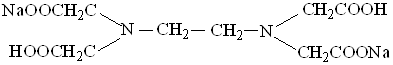
Препарат в виде белого растворимого в воде порошка является кристаллогидратом состава Nа2Н2Т · 2Н2О, где Т − анион ЭДТУ. ЭДТУ − слабая четырехосновная кислота (Н4Т), характеризующаяся четырьмя ступенями диссоциации: рКI = 2,0; рКII = 2,67; рКIII = 6,16; рКIV = 10,26. Близкие значения рКI и рКII показывают, что отщепление первых двух протонов протекает легче, чем отщепление двух оставшихся.
Данная соль относительно хорошо растворима в воде и диссоциирует, как сильный электролит:
Nа2Н2Т → 2Nа+ + Н2Т2−
Трилон Б взаимодействует со всеми катионами металлов II−IV аналитических групп. Реакции взаимодействия трилона Б с катионами металлов выражают уравнениями:
М2+ + Н2Т2−
![]() МТ2− + 2Н+
МТ2− + 2Н+
М3+ + Н2Т2−
![]() МТ− + 2Н+
МТ− + 2Н+
М4+ + Н2Т2−
![]() МТ + 2Н+
МТ + 2Н+
Таким образом, ЭДТА реагирует с ионами металлов с образованием комплекса 1:1, независимо от заряда катиона. В результате реакции взаимодействия происходит замещение двух катионов водорода, поэтому fэкв(М) = ½.
Указанные равновесия зависят от рН раствора. Для обеспечения полноты протекания реакции значение рН поддерживают постоянным, применяя для этого соответствующий буферный раствор. Чем выше заряд иона металла, тем прочнее образующийся комплексонат и тем более кислая среда допускается в ходе титрования.
Водные растворы комплексона III и большинства комплексонатов металлов бесцветны. Конец титрования обычно определяют с помощью металлиндикаторов. Это органические вещества, образующие цветные комплексные соединения с определяемым катионом. Свободный индикатор и его комплекс с ионом металла окрашены неодинаково. Устойчивость комплекса индикатора с металлом должна быть высокой, но ниже устойчивости комплекса металла с комплексоном.
Раствор трилона Б готовят из высушенной при 80°С навески. Для определения точной концентрации раствора трилон Б используют стандартный раствор МgSО4, приготовленный из стандарт-титра или навеску металлического цинка, растворенную в соляной или серной кислоте.
5.1. Стандартизация раствора трилон Б
Для определения точной концентрации раствора трилон Б применяется стандартный раствор сульфата магния, приготовленный из стандарт-титра. Концентрация раствора МgSО4 = 0,01 моль/л. В качестве индикатора используется кислотный хромовый темно-синий. В основе определения лежат реакции:
1) образование комплекса катиона магния с индикатором:
рН ~ 9
Мg2+ + Нind2−
![]() МgInd22−
+ 2Н+
МgInd22−
+ 2Н+
синий винно-красн.
2) образование более прочного комплекса катиона магния с рабочим раствором трилон Б с выделением свободной формы индикатора:
рН ~ 9
МgInd22−
+ Nа2Н2Т ![]() Nа2МgТ + 2Нind2−
Nа2МgТ + 2Нind2−
винно-красн. раб.р-р бесцв. синий
Методика анализа
В колбу для титрования последовательно из бюретки отмеряется 4,0; 5,0 и 6,0 мл стандартного 0,01 М раствора МgSО4, цилиндром отмеряется 5 мл аммиачного буферного раствора, добавляется 1 капля индикатора. Объем в колбе примерно вдвое разбавляется дистиллированной водой, смывая капли растворов со стенок внутрь колбы. Полученный раствор титруют трилоном Б до перехода винно-красной окраски раствора в синюю. Расчет концентрации раствора трилона Б производится на основе закона эквивалентов:
N(МgSО4) V(МgSО4)
N(Nа2Н2Т) = ---------
V(Nа2Н2Т)
По трем значениям концентрации раствора трилон Б рассчитывается среднее значение концентрации этого раствора.
Лабораторная работа №11
"Определение содержания Аl3+ методом обратного титрования"
Метод определения основан на том, что в кислой среде катионы алюминия образуют с трилоном Б прочное комплексное соединение (lg β = 16,13). Для разрушения гидратной оболочки комплекса и увеличения скорости реакции образования комплексоната алюминия проводят кипячение раствора соли алюминия с трилоном Б, взятом в избытке. По окончании реакции избыток трилона Б оттитровывается стандарным раствором сульфата цинка. В качестве индикатора используется ксиленоловый оранжевый, который в слабокислом растворе (рН ~ 5) в присутствии уротропина (гексаметилтетраамина) имеет лимонно-желтую окраску. От избыточной капли раствора сульфата цинка окраска индикатора меняется на красно-розовую за счет образования комплекса цинка с индикатором.
В основе определения лежат следующие реакции:
рН ~ 5
Аl3+ + Н2Т2−
![]() АlТ2− + 2Н+
+ Н2Т2−
АlТ2− + 2Н+
+ Н2Т2−
отмерен.изб. остаток
Н2Т2−
+ ZnSО4 ![]() ZnТ2− + 2Н+
ZnТ2− + 2Н+
остаток 2-ой раб.р-р
ZnSО4 + Нind2−
![]() ZnInd22− +
2Н+
ZnInd22− +
2Н+
изб.капля желт. красн.-роз.
По уравнению реакции и с учетом закона эквивалентов содержание алюминия определяется:
q(Аl3+) = q(Nа2Н2Т) − q(ZnSО4).
Методика анализа
В мерную колбу получают раствор задачи, доводят до метки дистиллированной водой и тщательно перемешивают. В колбу для титрования пипеткой переносят аликвотную часть задачи, добавляют из бюретки 10 мл рабочего раствора трилон Б и 3 мл 2 н раствора НNО3. Колбу накрывают стеклянной воронкой и раствор кипятят в течении 5 мин. Затем воронку над колбой ополаскивают дистиллированной водой, чтобы исключить потери раствора, и раствор охлаждают до комнатной температуры. В колбу для титрования добавляют 2 г предварительно взвешенного на технических весах уротропина. Содержимое колбы перемешивают круговыми движениями до растворения уротропина и добавляют на кончике шпателя ксиленоловый оранжевый. Смесь титруют вторым рабочим раствором ZnSО4 до перехода окраски индикатора из желтой в красно-розовую. Расчет содержания алюминия (III) в задаче проводят по среднему результату, полученному из данных титрования трех параллельных опытов:
N(Nа2Н2Т) · V(Nа2Н2Т) N(ZnSО4) · V(ZnSО4)
m(Аl3+) = 10 ·│----------------------------- − --------│· Мэ(Аl)
1000 1000
Лабораторная работа №12
"Определение общей жесткости воды"
Наличие Са2+ и Мg2+ в природных водах определяет их жесткость. Различают карбонатную (временную) и некарбонатную (постоянную). Первая обусловлена присутствием бикарбонатов (Са(НСО3)2 и Мg(НСО3)2), вторая − сульфатами (СаSО4 и МgSО4) или их хлоридами (СаСl2 и МgСl2). Карбонатную жесткость устраняют кипячением или прибавлением к воде гидроксида кальция:
Са(НСО3)2 + Са(ОН)2 → 2СаСО3↓ + 2Н2О
Постоянную жесткость устраняют прибавлением к воде соды:
СаSО4 + Nа2СО3 → СаСО3↓ + Nа2SО4
Сумма временной и постоянной жесткости воды составляет общую жесткость, выраженную мг-экв ионов Са2+ и Мg2+ на 1л воды.
В основе определения лежат реакции:
Мg2+ + Нind2−
![]() МgInd22−
+ 2Н+
МgInd22−
+ 2Н+
синий винно-красн.
Са2+ + Нind2−
![]() СаInd22−
+ 2Н+
СаInd22−
+ 2Н+
синий винно-красн.
рН ~ 9
(Са)МgInd22−
+ Nа2Н2Т ![]() Nа2(Са)МgТ + 2Нind2−
Nа2(Са)МgТ + 2Нind2−
винно-красн. раб.р-р бесцв. синий
Методика анализа
Пробу воды в мерной колбе доводят до метки дистиллированной водой и тщательно перемешивают. В колбу для титрования пипеткой отмеряют аликвотную часть исследуемого раствора, добавляют 5 мл аммиачного буферного раствора, разбавляют дистиллированной водой, смывая капли растворов со стенок внутрь колбы, и 1 каплю хром темно-синего.
Смесь титруют стандартным раствором трилона Б до перехода окраски индикатора из винно-красной в синюю. Определение повторяют трижды и вычисляют значение среднего объема трилона Б Vср(Nа2Н2Т), мл.
Результаты определения выражают суммарным количеством миллиграмм-эквивалентов (q) ионов кальция и магния в одном литре исследуемой пробы:
Vср(Nа2Н2Т) · N(Nа2Н2Т) · 1000
q = ----------------------------------------- ,
Vп
где Vп − объем пипетки, мл.
6. ПОТЕНЦИОМЕТРИЧЕСКИЙ МЕТОД АНАЛИЗА
|
|
1. Индикаторный стеклянный электрод 2. Насыщенный хлорсеребряный электрод 3. Исследуемый раствор 4. Магнитная мешалка 5. Магнитный стержень |
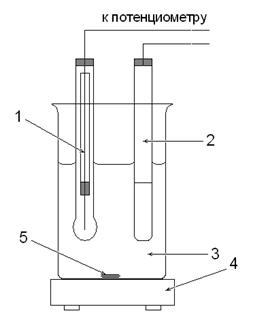
Рис. 1. Ячейка для определения рН раствора
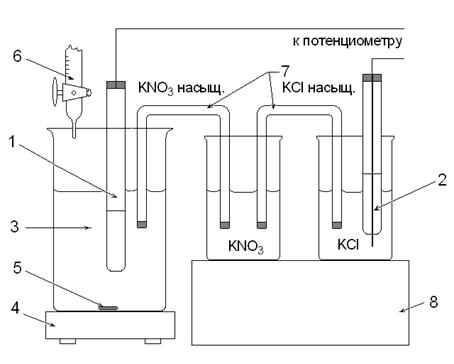
1. Индикаторный серебряный электрод. 2. Насыщенный хлорсеребряный электрод. 3. Исследуемый раствор. 4. Магнитная мешалка. 5. Магнитный стержень. 6. Бюретка с рабочим раствором AgNO3. 7. Мостики. 8. Штатив.
Рис. 2. Ячейка для определения галогенид-ионов.
Установка для потенциометрического титрования состоит из внешнего источника тока, чувствительного гальванометра (или постоянного усилителя), электролитической ячейки и регистрирующего прибора со шкалой в единицах рН или вольтах (см. рис. 1 и 2).
6.4. Правила работы на рН-метре-милливольтметре
В потенциометрическом титровании вместо потенциометров можно использовать рН-метры. рН-метр-милливольтметр типа рН-673М позволяет проводить анализ методом прямого потенциометрического титрования. Шкала прибора отградуирована в единицах рН или мВ и позволяет работать в диапазоне рН −1 ÷ 14 и в диапазоне потенциалов −1400 ÷ 1400 мВ.
Прибор готов к работе через 30 мин после включения в сеть.
Приступая к работе, необходимо тщательно промыть электроды дистиллированной водой, убедиться в правильности сборки электрохими-ческой цепи, а также в том, что электроды надежно закреплены и погружены в растворы, отрегулировать скорость вращения мешалки, чтобы исключить разбрызгивание раствора.
При малом объеме анализируемого раствора в стакан добавляют дистиллированную воду для того, чтобы концы электродов были погружены в раствор.
При отсчете показаний значений рН ориентировочные измерения проводят по нижней шкале прибора в диапазоне −1÷14.
На основании полученного предварительного значения рН точные измерения необходимо проводить в одном из узких диапазонов (−1÷4; 4÷9 или 9÷14), при этом отсчет проводят по верхней шкале.
При измерении потенциалов (+mV или −mV) также сначала ориентировочно оценивают величину потенциала по нижней шкале прибора, а при точном определении показаний по верхней выбранной шкале в соответствующем узком диапазоне, полученные значения умножают на 100.
6.5. Обработка результатов потенциометрического титрования
При проведении потенциометрического титрования по бюретке отмечают объем добавленного рабочего раствора (V, мл), а по шкале потенциометра (рН-метра-милливольтметра) полученные значения рН или потенциал (Е, мВ). Результаты титрования заносят в таблицу1 или 2.
Табл. 1
|
№ п/п V, мл рН ΔV, мл ΔрН ΔрН/ΔV V', мл |
| 1 2 3 4 5 6 7 |
|
1 0,00 2,06 2 0,20 2,10 0,20 0,04 0,08 0,10 3 0,40 2,15 0,20 0,05 0,10 0,30 4 0,60 2,22 0,20 0,07 0,14 0,50 … |
Табл. 2
|
№ п/п V, мл Е, мВ ΔV, мл ΔЕ, мВ ΔЕ/ΔV V', мл |
| 1 2 3 4 5 6 7 |
|
1 0,00 −206 2 0,20 −198 0,20 8 26,6 0,10 3 0,40 −188 0,20 10 33,3 0,30 4 0,60 −148 0,20 40 133,3 0,50 … |
В графу 2 таблицы записывают объем добавленного рабочего раствора (V, мл), в графу 3 − установившееся при этом значение рН или Е (мВ). В графы 4-7 записывают результаты обработки первичных экспериментальных данных. В графу 4 − разность объемов титранта между двумя последующими значениями 2-ой графы (шаг титрования); в графу 5 − разность показаний прибора между двумя последующими значениями графы 3; графа 6 − результат деления значения графы 5 на значение графы 4; в графу 7 (V', мл) помещают значения, рассчитанные как среднее арифметическое двух последующих значений графы 2.
Титрование считают законченным, когда после достижения максимальных значений ΔрН или ΔЕ прибавлено еще не менее трех порций рабочего раствора. По данным титрования строят на миллиметровой бумаге две кривые: в координатах "рН − V" или "Е − V" − интегральную кривую; в координатах " ΔрН/ΔV − V' " или " ΔЕ/ΔV − V' " − дифференциальную кривую. По дифференциальной кривой находят значение эквивалентного объема рабочего раствора (Vэкв, мл), которое используют для расчетов результатов анализа (рис.3).
Если в процессе титрования происходит последовательное титрование нескольких ионов (например, галогенидов раствором АgNО3), то на дифференциальной кривой будут наблюдаться несколько максимумов, каждому из которых будет соответствовать свой эквивалентный объем (рис.4). В этом случае предполагается, что титрование каждого последующего иона происходит лишь после полного осаждения предыдущего иона.
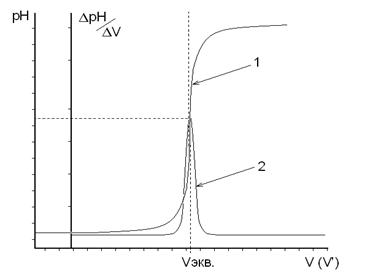
Рис. 3. Графики зависимости рН от объема добавленного титранта.
1 – Интегральная кривая 2 – Дифференциальная кривая
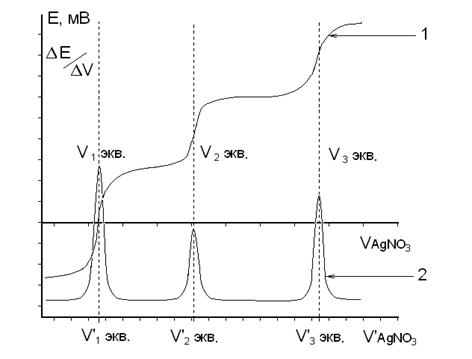
Рис. 4. Графики зависимости электродного потенциала от объема добавленного титранта.
1 – Интегральная кривая 2 – Дифференциальная кривая
ЛИТЕРАТУРА
1. Васильев В.П., Морозова Р.П., Кочергина Л.А. Практикум по аналитической химии. − М.: «Химия», 2000. − 327 с.
2. Пилипенко А.Т., Пятницкий И.В. Аналитическая химия. Т.I. − М.: «Химия», 1990. − 479 с.
3. Васильев В.П. Аналитическая химия. Т.I. − М.: «Высшая школа», 1989. − 319 с.
4. Алексеев В.Н. Количественный анализ. − М.: «Химия», 1972. −504 с.
5. Крешков А.П. Основы аналитической химии. Т.2. − М.: «Химия», 1976. − 480 с.